Viewpoint
Sep 2021
Peer-Reviewed

Many devices in current use were marketed before the US Food and Drug Administration (FDA) began regulating devices in 1976. Thus, manufacturers of these devices were not required to demonstrate safety and effectiveness, which presents both clinical and ethical problem for patients, especially for women, as some of the most dangerous devices—such as implanted contraceptive devices— are used only in women. This article investigates whether and to what extent devices for women receive less rigorous scrutiny than devices for men. This article also suggests how the FDA Center for Devices and Radiological Health could more effectively ensure safety and effectiveness of devices that were marketed prior to 1976.
The Center for Devices and Radiological Health (CDRH) of the US Food and Drug Administration (FDA) began regulating devices in 1976 after it was discovered that use of the Dalkon Shield and other women’s intrauterine contraceptive devices were linked to infertility.1 Congress and the FDA weighed the competing goals of providing the public reasonable assurance of safety and effectiveness and avoiding overregulation.1 Devices marketed prior to 1976 were grandfathered in with an amendment to the Food, Drug, and Cosmetic Act and were not required to establish safety and effectiveness.1 Such devices included mesh used for hernia repair, breast implants, and electroshock therapy devices. Currently, approximately 1% of devices enter the market through a process that generally requires clinical data, known as premarket approval (PMA). Approximately 30% of the remaining 99% of devices are “cleared” through the 510(k) pathway, which requires only that a device be “substantially equivalent” to a “predicate” device already on the market, without any requirement to demonstrate safety and effectiveness.2 Predicate devices, which can include those marketed before 1976,1 might never have received any FDA review for safety and effectiveness. Recalled devices have served as predicate devices, which raises ethical concerns, and even when a device has gone through trials, most subjects were men.3,4,5,6 This article specifically examines devices (ie, transcervical contraceptives, breast implants, vaginal meshes) intended for use in women. This article also suggests how the FDA Center for Devices and Radiological Health could more effectively ensure safety and effectiveness of devices that were marketed prior to 1976.
When a device fails a patient, it falls to the Division of Postmarket Surveillance at the FDA to identify patterns of problems with devices and then to formulate appropriate enforcement actions.7 These actions can vary from issuing or requiring issue of recalls, warning labels, or warning letters to manufacturers, organizations, or clinicians about safety concerns.8,9 The slow speed at which the FDA identifies patterns of harm has been under intense scrutiny, as the number of adverse events (AEs), ie, deaths, injuries, and malfunctions, reported to the FDA has doubled in the last 5 years—from 65 000 reports per month in 2016 to 115 000 reports per month in 2020 (see Figure).
Figure. Number of Adverse Event Reports for Medical Devices, 1995 Through May 2021a
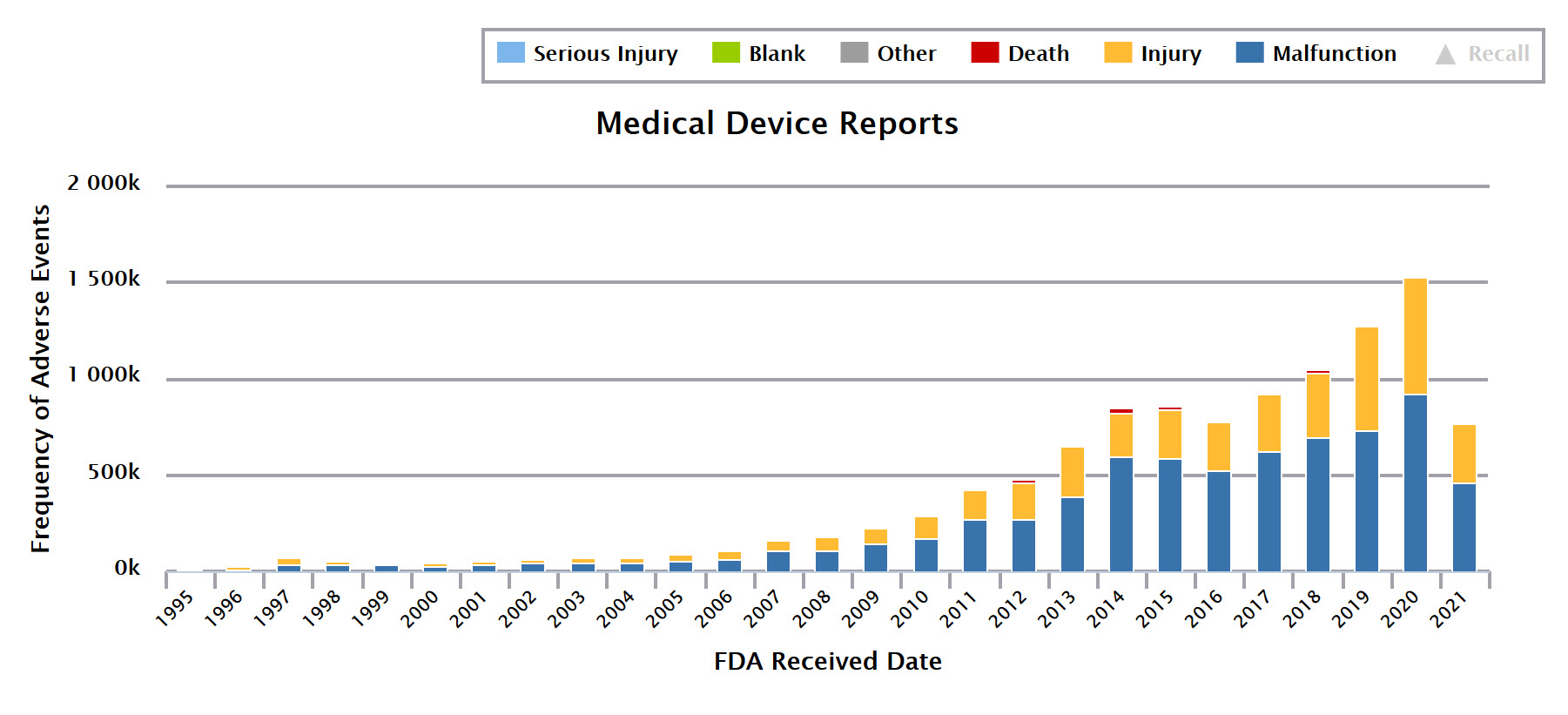
Abbreviation: FDA, Food and Drug Administration.
a Data from Device EventsTM database.10
The FDA’s current and most expeditious method of tracking problems with devices is via AE reporting by manufacturers and facilities, such as hospitals and laboratories. The Medical Device Reporting regulation requires a facility or manufacturer to self-report an adverse event within 30 days,11 or within 5 days if the “reportable event necessitates remedial action to prevent unreasonable risk of substantial harm to the public health.”12 When a facility contacts a device manufacturer to report a serious device-related concern, injury, or death, a manufacturer is mandated to file an AE report with the FDA.13 Of 11.4 million AE reports submitted to the FDA, most were reported from facilities via a manufacturer, 1.9 million were from physicians reported via a manufacturer; only 7291 were voluntarily submitted directly to CDRH by physicians.10
Attention to health problems caused by a transcervical contraceptive was generated by a popular film, The Bleeding Edge,14 and social media groups that encourage reporting AEs directly to the FDA rather than through a manufacturer. These groups recognized the need to directly report to the FDA when it was discovered that FDA AE reports were fewer in number than complaints submitted to manufacturers. For example, a social media group devoted to problems with a permanent birth control device had 22 000 members when the FDA held a public meeting on hysteroscopic sterilization devices in September 2015. Members suspected that the number of their complaints to manufacturers exceeded the 6000 AE reports shown in the FDA’s AE database at that time; Freedom of Information Act inquiries about FDA inspections of this device’s manufacturer yielded 2 inspection reports 3 years apart, with a combined 32 000 complaints that had not been submitted to the FDA by the manufacturer as AE reports. The FDA did not act until more severe events (ie, device migration and uterine perforation) were reported directly to the agency by patients; the company settled the claims for $1.6 billion.15
Adverse event reports data are critical for patients trying to exercise self-determination and to make informed decisions.
However, the FDA public meeting on hysteroscopic sterilization devices generated publicity. One US Congressman questioned the FDA about the device’s safety.16 Physicians became more aware of links between the device and their patients’ health, resulting in submission of another 5400 reports to the FDA with the reporter occupation designated as “physician.”10 Although the FDA required a “black box” warning to be added to the device’s packaging in February 2016 and ordered the manufacturer to conduct a new clinical study on risks,17 it did not mandate or recommend a recall, offering the reason that the agency did not want to limit patients’ choices. After 16 years on the market, the manufacturer voluntarily withdrew the device from international commercial distribution in September 2017, citing “commercial reasons”18 and then domestically in December 2018,19 following release of The Bleeding Edge, which documented the trauma and dangers of this device in women.14 So, why is the FDA slow to ensure that women’s devices are safe and effective?
Consider another example. Scrutiny of breast implant material in the late 1980s prompted a moratorium on silicone breast implants from 1992 to 2006,20 yet lack of transparency about AE reports resulted in patients and physicians assuming, for 20 years, that newer breast implant materials were safer than those targeted by the moratorium. Again, because women harmed by breast implants spoke up, search for AE reports ensued. How could there be more than 100 000 members in breast implant social media support groups and so few AE reports?
Patient safety advocates began meeting with the FDA in 2015; in 2018, the agency revealed that the adverse event reports existed but were not made public, despite the mandatory reporting requirement. During a public meeting in 2019, the FDA disclosed receipt of more than 300 000 breast implant AE reports—more than 20 times the number made public.21 After this meeting, then-FDA Commissioner Scott Gottlieb promised release of all AE reports collected through “alternative summary reporting,”22 most of which were released in June 2019.23
Only 54 400 of 5.8 million alternative summary reports made public by the FDA in June 2019 were for devices or materials marketed solely to men. Vaginal mesh AE reports have still not been released. It is not known how many of these reports exist, and I argue here that withholding them during pending legal cases is unethical, since members of the public—who have been or could be harmed by a device or material—need access to this data, which is intended by Congress to be publicly available. This data is critical for patients trying to exercise self-determination and to make informed decisions about whether and when to have a device or material implanted or about whether and how to respond to a past decision made without access to existing information that should have been public and could have informed their risk-benefit analyses prior to surgical implantation. Sharing the actual number of AEs and their timeline is critical for public protection.
In addition to reviewing AEs, CDRH monitors devices’ postmarket safety and effectiveness. Although AEs are public, more than 1 million reports are made available per year in the Manufacturer and User Facility Device Experience Database, and the FDA does not release them in a format that is easy to review or understand. The FDA does not have resources to proactively identify device problems, and even when the agency orders postmarket surveillance studies, it can struggle to enforce manufacturers’ compliance.24
For example, postmarket surveillance studies of all major brands of breast implants were ordered by the FDA in 2006 and remain incomplete. The FDA updated the study parameters in 2013 and 2014 to allow for smaller studies, since the manufacturers struggled with enrollment and patient follow-up. Every 1 to 2 years, the CDRH sends warning letters to the breast implant manufacturers, who respond with requests for additional time to complete these studies. After each request, the FDA allows additional time, seemingly due to lack of resources or mechanisms for compliance enforcement. Despite warning that delay and noncompliance can compromise device marketing and distribution, no further enforcement (ie, production suspension, moratorium on sales, withdrawal from the market, or recall) has been enacted, rendering the requirement for postmarket surveillance meaningless.
Ethical and clinical questions about withdrawing a device from the market arise when no alternative exists: Are patients safer when a device of questionable or unknown safety and effectiveness is at least available rather than unavailable? Breast cancer survivors, for example, express concern about limited breast implant material options. Is a 2020 FDA-recommended black box warning for breast implants25 best regarded, ethically and clinically, as promoting safety or as generating risk awareness that further limits an already narrow set of options? Even with a warning in place, a patient is not the one who actually removes packaging for an implant, so will she see the black box warning? Should she see it? How many adverse events should be regarded as too many? As Jeanne Lenzer noted: “The question is not whether a device ever causes harm but whether the benefits are expected to exceed the harm in a defined population.”26
The FDA plans to more proactively identify devices’ risks, but these initiatives are years from implementation, as they require integrating health care organizations' electronic health records (EHRs) and must be adopted by clinicians and payers. The CDRH Health of Women Initiative, launched in December 2016,27 for example, focuses on device performance across women’s lifespans and includes tracking device use via claims payments systems by 2023 and future tracking of adverse events via EHRs—once the EHR vendors are in full compliance.
It is essential to make the mechanism for AE reporting user friendly and widely accessible and to ensure transparency of AE reporting, as well as enforce requirements for postmarket surveillance to protect women, as well as men, from having dangerous devices implanted without knowledge of potential harms and benefits.
Institute of Medicine. Medical Devices and the Public’s Health: The FDA 510(k) Clearance Process at 35 Years. National Academies Press; 2011.
Sullivan T. Institute of Medicine report Medical Devices and the Public’s Health: The FDA 510(k) Clearance Process at 35 Years. Policy and Medicine. May 6, 2018. Accessed December 30, 2020. https://www.policymed.com/2011/07/institute-of-medicine-report-medical-devices-and-the-publics-health-the-fda-510k-clearance-process-a.html
Institute of Medicine. Safe Medical Devices for Children. National Academies Press; 2006.
CDRH transparency: compliance and enforcement. US Food and Drug Administration. Updated September 7, 2018. Accessed June 2, 2021. https://www.fda.gov/about-fda/cdrh-transparency/cdrh-transparency-compliance-enforcement
Letters to healthcare providers. US Food and Drug Administration. Updated May 27, 2021. Accessed June 2, 2021. https://www.fda.gov/medical-devices/medical-device-safety/letters-health-care-providers
Device Events™ database. Proprietary. Device Events, LLC; 2015.
Mandatory reporting requirements: manufacturers, importers and device user facilities. US Food and Drug Administration. Updated May 22, 2020. Accessed June 2, 2021. https://www.fda.gov/medical-devices/postmarket-requirements-devices/mandatory-reporting-requirements-manufacturers-importers-and-device-user-facilities
21 CFR §803.53 (2021).
Medical device reporting (MDR): how to report medical device problems. US Food and Drug Administration. Accessed February 25, 2021. https://www.fda.gov/medical-devices/medical-device-safety/medical-device-reporting-mdr-how-report-medical-device-problems
Dick K. The Bleeding Edge. Netflix; 2018. Accessed March 1, 2021. https://www.netflix.com/title/80170862
Randazzo S. Bayer settles Essure birth control litigation for $1.6 billion. Wall Street Journal. August 20, 2020. Accessed June 2, 2021. https://www.wsj.com/articles/bayer-settles-essure-birth-control-litigation-for-1-6-billion-11597954204
Belleisle JW. Congressman Mike Fitzpatrick questions FDA about Essure reporting changes. Legal Reader. April 21, 2016. Accessed June 2, 2021. https://www.legalreader.com/congressman-mike-fitzpatrick-questions-fda-about-essure-reporting-changes/
Thompson D. FDA orders “black box” warning label on Essure. WebMD. February 29, 2016. Accessed June 2, 2021. https://www.webmd.com/sex/birth-control/news/20160229/fda-orders-black-box-warning-label-on-essure-long-acting-contraceptive#:~:text=MONDAY%2C%20Feb.,and%20Drug%20Administration%20announced%20Monday
McGinley L. Sales of Essure birth control implant to be halted by Bayer; US last to sell controversial device. Washington Post. July 20, 2018. Accessed July 9, 2021. https://www.washingtonpost.com/news/to-your-health/wp/2018/07/20/sales-of-essure-birth-control-implant-halted-by-bayer-u-s-was-last-to-sell-controversial-device/
FDA in brief: FDA provides updates on discontinuation of Essure and ongoing postmarket activities. US Food and Drug Administration. January 10, 2020. Accessed April 19, 2021. https://www.fda.gov/news-events/fda-brief/fda-brief-fda-provides-updates-discontinuation-essure-and-ongoing-postmarket-activities#:~:text=The%20manufacturer%2C%20Bayer%2C%20voluntarily%20stopped,unused%20Essure%20devices%20to%20Bayer
Aspan M. “They killed her”: why are breast implants still putting millions of women at risk? Fortune. May 18, 2020. Accessed April 19, 2021. https://fortune.com/longform/breast-implants-dangerous-allergan-abbvie-acquisition/?j16sc8
Densford F. FDA head Gottleib pledges to release all adverse event reports. MassDevice. March 28, 2019. Accessed June 2, 2021. https://www.massdevice.com/fda-head-gottlieb-pledges-to-release-all-adverse-event-reports/
Hallman B. FDA releases vast trove of hidden medical device injury and malfunction reports. International Consortium of Investigative Journalists. June 24, 2019. Accessed June 2, 2021. https://www.icij.org/investigations/implant-files/fda-releases-vast-trove-of-hidden-medical-device-injury-and-malfunction-reports/
US Food and Drug Administration. Breast implants—certain labeling recommendations to improve patient communication. Guidance for industry and Food and Drug Administration staff. September 29, 2020. Accessed February 25, 2021. https://www.fda.gov/media/131885/download
Lenzer J. The Danger Within Us: America’s Untested, Unregulated Medical Device Industry and One Man’s Battle to Survive It. Little, Brown & Co; 2017.
CDRH Health of Women Program. US Food and Drug Administration. Reviewed September 10, 2020. Accessed October 30, 2020. https://www.fda.gov/about-fda/center-devices-and-radiological-health/cdrh-health-women-program



